 2016, Vol. 37
2016, Vol. 37



2. 中国林业科学研究院 热带林业研究所,广东 广州 510520;
3. 广东科技学院,广东 东莞 523083
2. Research Institute of Tropical Forestry, Chinese Academy of Forestry, Guangzhou 510520, China;
3. Guangdong University of Science and Technology, Dongguan 523083, China
单核苷酸多态性(Single nucleotide polymorphism,SNP)是指生物体基因组中单个碱基的颠换或转换导致的DNA变异,是最普遍和最广泛的基因组变异形式,如拟南芥Arabidopsis thaliana 3个品系间存在超过82万个SNP,平均约1.4 kb就存在1个SNP[1]。SNP目前已广泛应用于生物、医学和农学等研究领域[2-3]。
传统的PCR片段测序中,测序厂家所提供的软件只能识别各波峰位置的单个碱基,而对二倍体个体内存在SNP的双峰处的低峰碱基不能有效检测。已有一些第三方软件可用于二倍体物种PCR片段测序的SNP自动检测,如Mutation Surveyor (http://www.softgenetics.com/MutationSurveyor.html)、novoSNP[4]和PolyPhred[5]等。但是,这些软件均需参考序列,具有局限性,不能有效用于一些序列的分析,如表达序列标签(Expressed sequence tag,EST)和候选基因的测序中的内含子区域,而且操作上较为繁琐。这些软件的检测准确性均偏低[6]。此外,PolyPhred[5]还需要至少8个测序文件以保证足够的准确性,不利于少量样品的检测。针对二倍体PCR片段测序中如何进行SNP的自动、有效检测,本文基于模式识别的方法构建了无需参考序列进行SNP自动检测的计算机系统,并验证了系统的准确性。
1 材料与方法 1.1 系统开发平台本研究基于LabWindows/CVI (以下简称CVI)平台,以CVI 9.0作为系统前台进行主界面的设计和基本功能菜单的实现。CVI是基于C语言的虚拟仪器软件开发平台,其功能强大,具有灵活的交互式编程方法和丰富的用户接口资源[7-8],且运行速度快、界面控件丰富。
采用MATLAB 2012函数库编程实现模式识别的分类器构建。MATLAB具有编程简单、仿真能力强和易于扩展移植等优点[9],提供内部神经网络工具箱,且可外接用户自行开发的模式识别算法或软件包,有利于用户进行各种模式识别算法的测试与建模。本文主要通过MATLAB实现模式识别的分类器结构的建立,利用反向传播神经网络(Back-propagation neural network, BPNN)进行权值和阈值的训练,再将训练好的BPNN结构移植至CVI环境用作SNP分类器。
系统的运行环境为Windows XP Professional、Windows 7或Window 8。
1.2 系统功能模块和界面的设计系统主要包括测序数据导入、碱基分离、噪声处理、SNP识别、数据显示与存储、人工校正等功能模块。测序数据导入可打开扩展名为.ab1或.scf的测序文件。碱基分离是根据测序数据中G、A、T和C 4种碱基的不同荧光颜色而将它们分别提取出来。噪声处理通过一维离散小波变换滤除噪声,便于后续的数据特征提取。SNP识别主要根据测序峰图的波形特征,结合二倍体内SNP表现为双峰的现象,提取各碱基位置的典型波形特征作为BPNN的输入向量,输出SNP属性分数,再参照判别标准给出SNP等级。数据显示可在运行窗口直观显示测序峰图和序列以及SNP的位置、双峰碱基、属性分数和等级,识别结果可存为.txt文件。人工校正允许用户对误判和漏判的SNP进行删除、添加和更改碱基等手动操作。系统的工作流程见图 1。

|
图 1 SNP检测流程图 Figure 1 A flow chart of the SNP detection process |
系统界面包括主操作区、原始峰图显示区、SNP识别结果区和人工校正区等4个功能区域(图 2)。

|
图 2 SNP检测系统的界面 Figure 2 A typical interface of the SNP detection system |
主操作区位于系统界面的上部,如图 2所示,包含面板选项卡、菜单栏、快捷键和信息提示栏。面板选项卡可提供多个检测面板,各检测面板的序列分析数据相互独立,以便进行多个测序文件的检测。菜单栏包括文件、编辑、滤波选择、识别方法和帮助等一级菜单,各一级菜单可下拉显示二级菜单。文件的二级菜单包括原始测序文件导入、SNP检测结果保存等。编辑的二级菜单包括X轴和Y轴缩放以及隐藏/显示碱基G、A、T和C。滤波选择的二级菜单包括小波变换等方法。识别方法的二级菜单包括BPNN等多种方法。快捷键工具栏列出常用功能,包括文件打开、SNP检测(按默认的滤波和模式识别方法)、增加面板、关闭当前面板、峰图和结果显示窗口X轴和Y轴缩放等。信息提示栏显示文件路径等。
原始峰图显示区位于界面右中区域,见图 2,不同颜色代表碱基G、A、T和C显示测序的峰图,可通过主操作区的快捷按钮选择性地显示一种或几种碱基类型,也可进行X轴和Y轴的缩放。
SNP识别结果区位于界面下部区域,见图 2,界面右下区域显示SNP识别结果,包括碱基位置、碱基类型、双峰SNP、SNP分数和SNP等级等。界面左下区域列表总结SNP检测的结果,点击某一行可与右下窗口相应位置的SNP进行快速定位。
人工校正区位于界面左中区域(图 2),允许用户对任一位置碱基进行更改、删除和增加等操作。
1.3 系统主要功能的实现 1.3.1 测序数据的导入和碱基分离本系统可导入.ab1或.scf格式的测序文件,根据文件内容分离出各种碱基。以.ab1文件为例说明。按照美国Applied Biosystems公司的ab1f文件格式,.ab1文件是由一个指定的文件夹存储文件信息,包括文件名称、碱基类型和碱基数量等。文件7 ~ 34字节是‘DirEntry’结构体,指向文件夹所在的位置,其结构为:
Struct DirEntry{ sInt32 name; sInt32 number; sInt16 elementtype; sInt16 elementsize; sInt32 numelements; sInt32 datasize; sInt32 dataoffset; sInt32 datahandle; },
其中,number显示通道编号(对应4种碱基),elementsize显示碱基所占空间大小,numelements显示碱基数量,dataoffset显示碱基位置的偏移量。根据这些信息即可将各通道的碱基和荧光数据提取出来。
1.3.2 滤波的实现分离出的碱基荧光信号通常含有噪声,影响SNP分析的准确性,可通过小波分解进行噪声滤除。小波分解具有良好的时频分析特性,在时域和频域内都具有突显信号局部特征的能力,并且能够在多种分辨率下观测数据,是噪声滤除的有效方法[10-11]。
本系统调用了CVI中Signal Processing Toolkit 7.0.2工具箱所含的小波工具包的函数,一维离散小波的滤波过程如下:分离4个通道的碱基数据; 读取各通道的碱基数据长度,获得滤波参数; 调用SptAllocCoeffWFBD函数,分配小波滤波工具包的滤波参数结构; 调用SptReadCoeffWFBD函数,读取小波工具包参数; 调用SptDiscreteWaveletTransform (filterData, dataLenght, analysisFilter, SptSymmetric, NULL, NULL, scales, shift, dwt, &outputSize, lengths)函数,进行一维离散小波分解,滤除后的数据存储于变量dwt中,用于后续分析。
1.3.3 SNP识别分类器的构建利用BPNN构建SNP识别分类器。BPNN是一种反向传播且能修正误差的多层映射模式,由输入层、隐含层和输出层及各层神经元之间连接组成[12]。SNP识别可以视作从波形特征输入到SNP类型输出的非线性映射问题。
基于文献[13],选择了SNP处双峰的波峰距离、高度比值和起伏度比值作为特征参数,归一化处理后作为BPNN输入层的特征向量,即输入层的神经元数为3。BPNN输出层为SNP类别的诊断结果,可能是双峰SNP位点或单峰非SNP位点,即输出层神经元为1;输出层中设定范围0 ~ 100为1个位点的属性分数,并结合属性分数和周边杂峰将SNP可能性分为6个等级,等级越小越可能为SNP。
MATLAB神经网络工具箱中调用BPNN构建SNP分类器的过程具体如下:调用net=newff[input, output, 10, 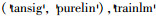
因SNP检测可能存在假阳性和假阴性,人工校正的碱基删除、添加和更改的结果可直接显示在当前面板。SNP检测结果的保存格式为.txt文件。
利用尾叶桉Eucalyptus urophylla 26个测序文件的143个SNP进行系统测试,并与Mutation Surveyor (http://www.softgenetics.com/MutationSurveyor.html)、novoSNP[4]和PolyPhred[5]的结果进行比较,分析软件的有效性。所用26个测序文件为尾叶桉SNP开发的EST重测序文件[14]。
2 结果与分析 2.1 系统主要功能的实现系统导入测序文件后能正确地进行碱基判读,判读结果与测序结果较为一致。图 3显示了本系统对1个序列的判读与测序结果一致。但对测序质量较差或者杂峰较多的区域,测序结果亦不准确,与本系统的判读结果可能有异。图 3B表明本系统能可靠地识别双峰碱基位置的SNP,155 bp处的SNP判读为T/A,分值为87,等级为1。并且,其他较低的杂峰被有效排除,未被误判为SNP。

|
图 3 本系统导入测序文件后的判读序列与测序比较 Figure 3 Base sequence obtained in our system being compared to that of sequencer b:本系统判读的序列,155 bp处双峰、SNP为T/A |
人工校正可对特定位置的第1和/或第2碱基进行修改、删除和添加操作。图 4显示了对174 bp处误读的‘T’修改为‘G’,表明系统可有效地进行人工校正。

|
图 4 人工校正的有效性 Figure 4 Efficient manual correction of a misidentified base |
数据存储的.txt文件见图 5,第1行为文件名,数字代表显示碱基位置,识别序列上面一行为碱基序列,下面一行显示识别的SNP的第2碱基。测序前20个碱基一般是较杂乱的峰,SNP可靠性低。

|
图 5 SNP检测结果存为.txt文件 Figure 5 SNP detection result saved as a.txt file |
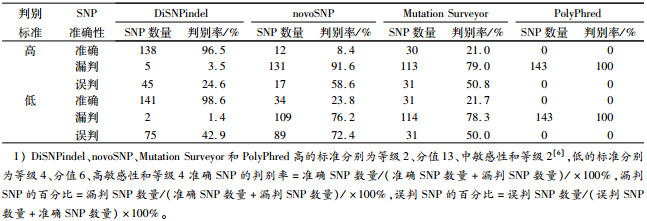
对于测试的尾叶桉26个测序文件,对比另外3个软件,本系统具有较高的准确性和误检率。表 1显示了不同软件在高和低的标准下对143个SNP的识别准确率、漏判(假阴性)率和误判(假阳性)率,不管是在高还是低的标准下,本文所开发的系统均有最高的SNP判读准确率以及最低的假阴性率和假阳性率,PolyPhred除外,因其未检测到任何SNP。
|
|
表 1 不同软件识别SNP的结果对比1) Table 1 Comparison of software performance in SNP detection |
本文基于CVI 9.0开发平台,结合MATLAB函数库,运用特征提取和模式识别等技术构建了二倍体片段测序的SNP自动检测系统。本系统开发中结合了CVI运行速度快和用户接口资源丰富以及MATLAB函数库多样和用户可自行扩充函数等优点,降低了系统开发的难度,并且检测系统界面友好、易于扩充。系统各模块功能的完整实现和测试的有效性也证明相关技术的合理运用。
高通量的新一代测序技术已经兴起,但传统的PCR片段测序仍非常重要[15]。测序厂家提供的软件只能识别各序列位置的最高峰所对应的碱基,因此双峰位置的低峰碱基的识别需要第三方软件。另外,测序也常出现杂峰,且杂峰可能高于碱基峰而导致测序厂家的软件不能正确读序。本文针对这一问题研发了SNP自动检测系统,可以有效实现双峰判读和杂峰滤除等功能,为二倍体PCR片段测序的SNP判读提供了有力工具。
与功能类似的软件Mutation Surveyor (http://www.softgenetics.com/MutationSurveyor.html)、novoSNP[4]和PolyPhred[5]相比,本系统不需参考序列,既为EST和候选基因的测序中内含子无参考序列的SNP检测提供了便利,也减少了输入参考序列的繁琐操作; 并且,本文所开发的检测系统具有最高的SNP判读准确率以及最低的假阴性率和假阳性率,可用于二倍体PCR片段测序中SNP的高效检测。
实际应用中,Mutation Surveyor和novoSNP可能假阳性率极高,如对15个基因171个SNP的检测中的假阳性SNP分别为3 728和505个,显著多于正确判读的SNP[16]。另一方面,任何检测系统的准确率都严重依赖于测序质量,实际应用时需要优化测序体系[4]。
| [1] |
OSSOWSKI S, SCHNEEBERGER K, CLARK R M, et al. Sequencing of natural strains of Arabidopsis thaliana wit short reads[J]. Genome Res, 2008, 18(12): 2024-2033. DOI:10.1101/gr.080200.108 (  0) 0) |
| [2] |
唐立琼, 肖层林, 王伟平. SNP分子标记的研究及其应用进展[J]. 中国农学通报, 2012, 28(12): 154-158. ( 0) |
| [3] |
许家磊, 王宇, 后猛, 等. SNP检测方法的研究进展[J]. 分子植物育种, 2015, 13(2): 475-482. ( 0) |
| [4] |
WECKX S, DEL-FAVERO J, RADEMAKERS R, et al. novoSNP, a novel computational tool for sequence variation discovery[J]. Genome Res, 2005, 15(3): 436-442. DOI:10.1101/gr.2754005 ( 0) |
| [5] |
MATTHEW S, JAMES S, ROBERTSON P D, et al. Automating sequence-based detection and genotyping of single nucleotide polymorphisms (SNPs) from diploid samples[J]. Nat Genet, 2006, 38(3): 375-381. DOI:10.1038/ng1746 ( 0) |
| [6] |
DENG J Z, HUANG H S, YU X L, et al. DiSNPindel: Improved intra-individual SNP and InDel detection in direct amplicon sequencing of a diploid[J]. BMC Bioinformatics, 2015, 16: 343. DOI:10.1186/s12859-015-0790-y ( 0) |
| [7] |
仇志平, 李树军. LabWindows/CVI虚拟仪器软件在测试领域中的应用[J]. 计算机工程与设计, 2007, 28(22): 5544-5548. DOI:10.3969/j.issn.1000-7024.2007.22.065 ( 0) |
| [8] |
刘君华. 虚拟程序编程语言LabWindows/CVI编程[M]. 北京: 电子工业出版社, 2001.
( 0) |
| [9] |
肖伟, 刘忠, 曾新勇, 等. MATLAB程序设计与应用[M]. 北京: 清华大学出版社, 2005.
( 0) |
| [10] |
BUI T D, CHEN G. Translation-invariant denoising using multiwavelets[J]. IEEE Trans Sig Proc, 1998, 46(12): 3414-3420. DOI:10.1109/78.735315 ( 0) |
| [11] |
PAN Q, ZHANG P, DAI G, et al. Two denoising methods by wavelet transform[J]. IEEE Trans Sig Proc, 1999, 47(12): 3401-3406. DOI:10.1109/78.806084 ( 0) |
| [12] |
MCKEOWN J J, STELLA F, HALL G. Some numerical aspects of the training problem for feed-forward neural nets[J]. Neural Netw, 1997, 10(9): 1455-1463. ( 0) |
| [13] |
黄华盛. 基于模式识别的二倍体个体内SNP和InDel自动检测[D]. 广州: 华南农业大学, 2014.
( 0) |
| [14] |
YU X, GUO Y, ZHANG X, et al. Integration of EST-CAPS markers into genetic maps of and Eucalgptus urophylla and E.tereticornis and their alignment with E. grandis genome sequence[J]. Silvae Genet, 2012, 61(6): 247-255. ( 0) |
| [15] |
STUDER A, ZHAO Q, ROSS-IBARRA J, et al. Identification of a functional transposon insertion in the maize domestication gene tb1[J]. Nat Genet, 2011, 43(11): 1160-1163. DOI:10.1038/ng.942 ( 0) |
| [16] |
NGAMPHIW C, KULAWONGANUNCHAI S, ASSAWAMAKIN A, et al. VarDetect: A nucleotide sequence variation exploratory tool[J]. BMC Bioinformatics, 2008, 9(S12): 9. ( 0) |